
You don’t have to be a scientist to do science.
By simply running a free program, you can help advance research in medicine, clean energy, and materials science.
|
|
How does it work?
By running Rosetta@home on your computer when you're not using it you will speed up and extend our efforts to design new proteins and to predict their 3-dimensional shapes. Proteins are the molecular machines and building blocks of life. You can read more about protein folding and design here.
Follow us on Twitter: @rosettaathome
Rosetta@home is not for profit.
Join Ralph@home to help improve this project.
User of the Day


Predictor of the day
Predictor of the day: Congratulations to 2buiy5RiX3bN8JBr2yuiTCdV5yLJ (Team Gridcoin) for predicting the lowest energy structure for workunit rb_02_26_22117....
1 Mar 2022, 0:00:00 UTC
· Discuss
... more
Predictor of the day is available as an RSS feed
| As of 17 May 2026, 7:22:25 UTC [ Scheduler running ] | |
| Total queued jobs: | 0 |
| In progress: | 9 |
| Successes last 24h: | 34 |
Users  (last day ): (last day ): |
1,392,336 (+1) |
| Hosts (last day ): |
4,578,598 (+1) |
| Credits last 24h : |
71,745 |
| Total credits : |
162,234,502,919 |
| TeraFLOPS estimate: | 0.717 |
News
Foldit on the Web!
Introducing Foldit Education Mode v2 on the Web
In 2020, as the pandemic set in and classes moved online, the Foldit team realized they had a tremendous opportunity to help the science education community that was desperate for good science learning tools. They therefore created and released Foldit Education Mode in August 2020 as a ready-made teaching tool for biochemistry that instructors could use easily and freely in their courses. In the past five years, at least hundreds of educators and many thousands of students have used Foldit Education Mode.
While Foldit Education Mode was greatly successful at meeting the challenge of the moment, it still had flaws that would hold some students back from using it. The Foldit team also recently published a quantitative assessment of learning with Foldit Education Mode (https://doi.org/10.1002/bmb.21906), and while it was fulfilling its purpose, there was definitely also room to improve.
That’s why they're very excited to introduce their second release of Foldit Education Mode with improved levels and teaching tools, but even more excitingly: now available on the web! This means no more downloads required, and compatibility with any computer type. For many, this will enable use of Foldit in the classroom that was never previously possible, and for many more a great degree of increased flexibility and improved user experience.
To play Foldit Education Mode on the web, please go to https://play.fold.it/, and you can start playing immediately! You’ll notice several updates to the user interface along with the new and improved puzzles. The number of puzzles is slightly larger, but this change was largely made to break up levels that had too much biochemistry and gameplay content all rolled together, and by separating it out, the learning experience becomes considerably easier.
In the long term, the Foldit team plans to explore the possibility of hosting smaller Science puzzles to help bridge the gap for new players who start with the web version, but that will require more work to complete. In the meantime, they hope that educators and students worldwide will enjoy the new and improved experience, and those who couldn’t participate before will now be able to.
As always, the Foldit team would appreciate finding out who is using Foldit Education Mode, as they don’t collect that data; it really helps with procuring more funding to support these efforts. So if you do use it, please drop them a line at mail.fold.it@gmail.com.
This post originated from the Foldit blog post https://fold.it/forum/blog/introducing-foldit-education-mode-v2-on-the-web
11 Sep 2025, 22:35:26 UTC
· Discuss
Rosetta@home Update
First, we’d like to thank all our contributors who have supported and continue to contribute to our scientific projects through Rosetta@home. We want to update you on the status of Rosetta@home projects and our future plans.
With the advancement of AI models like AlphaFold and RosettaFold for protein structure predictions, Rosetta@home has been less used for this purpose. However, researchers are now utilizing Rosetta@home for small molecule and peptide designs, where even the current state-of-the-art AI models struggle due to limitations in generalizability to novel small molecules and non-canonical peptides.
Recently, we have developed a virtual screening protocol in Rosetta, named RosettaVS, for small molecule drug discovery. This work has been published in Nature Communications (https://doi.org/10.1038/s41467-024-52061-7), demonstrating that RosettaVS is one of the best physics-based virtual screening protocols. Combined with deep learning techniques, it can effectively screen multi-billion compound libraries and discover novel compounds for pharmaceutical targets.
While deep learning models like AlphaFold and RosettaFold can predict canonical peptide structures, they cannot handle peptides with non-canonical amino acids or mixed chirality. The physics-based force field in Rosetta has specialized terms to simulate these amino acids. Rosetta will be used to sample hundreds of thousands of different conformations of the designed peptide to validate the structure.
Looking ahead, Rosetta@home will be an invaluable platform for large-scale virtual screening and peptide simulations for drug discovery. We plan to launch more virtual screening jobs and peptide simulations on Rosetta@home in the near future.
Thank you!
4 Mar 2025, 4:50:22 UTC
· Discuss
COVID-19 vaccine with IPD nanoparticles wins full approval abroad
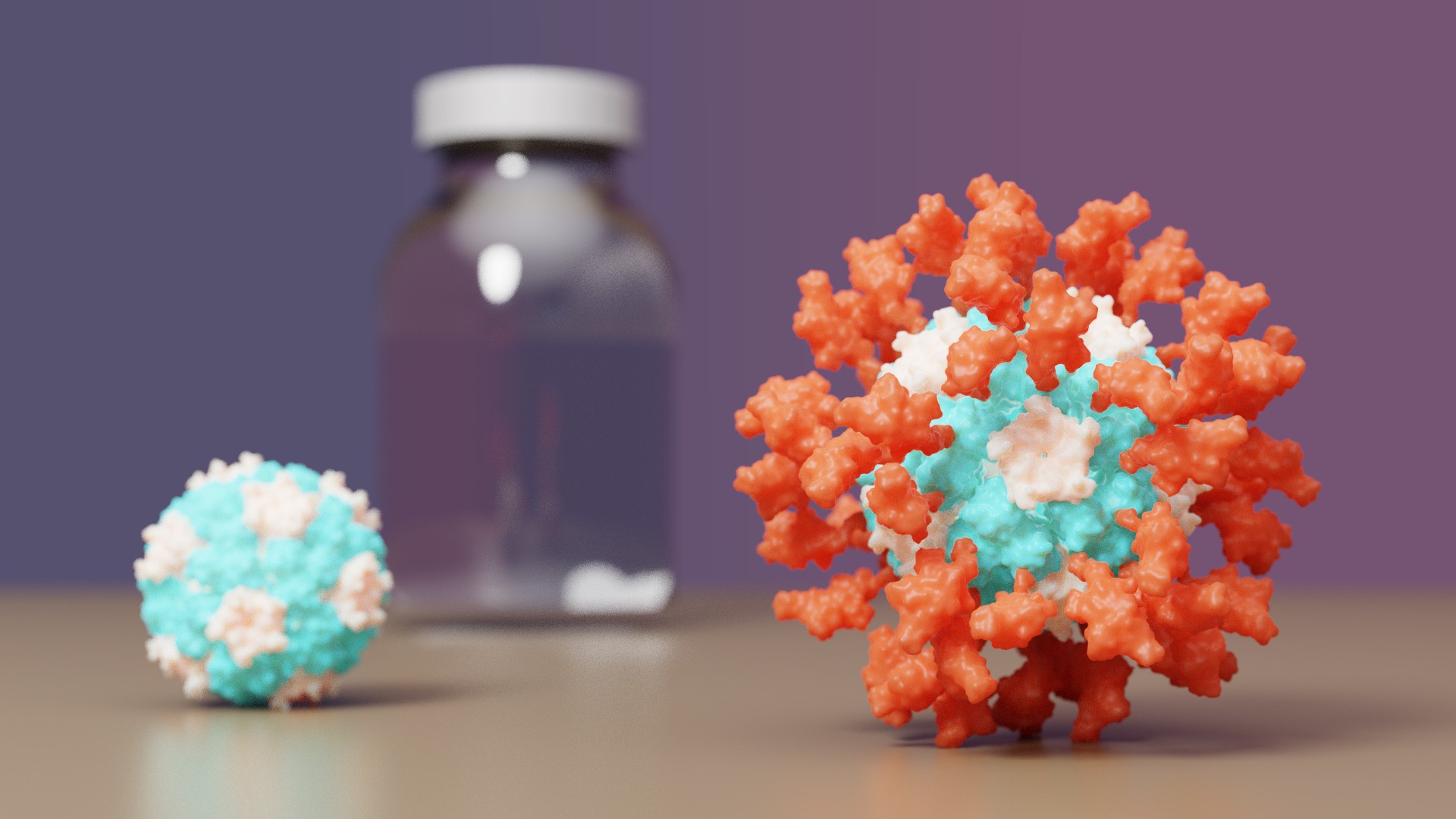
The IPD is excited to announce it's first designed protein medicine with full approval abroad.
Congrats and thank you to all Rosetta@home contributors! The computing you have provided has greatly aided in de novo protein design challenges such as vaccine development leading to breakthroughs like this.
For more information you can visit the IPD vaccine news post.
A video is also available here.
From the IPD news site:
• Clinical testing found the vaccine outperforms Oxford/AstraZeneca’s
• The protein-based vaccine, now called SKYCovione, does not require deep freezing
• University of Washington to waive royalty fees for the duration of the pandemic
• South Korea to purchase 10 million doses for domestic use
A vaccine for COVID-19 developed at the University of Washington School of Medicine has been approved by the Korean Ministry of Food and Drug Safety for use in individuals 18 years of age and older. The vaccine, now known as SKYCovione, was found to be more effective than the Oxford/AstraZeneca vaccine sold under the brand names Covishield and Vaxzevria.
SK bioscience, the company leading the SKYCovione’s clinical development abroad, is now seeking approval for its use in the United Kingdom and beyond. If approved by the World Health Organization, the vaccine will be made available through COVAX, an international effort to equitably distribute COVID-19 vaccines around the world. In addition, the South Korean government has agreed to purchase 10 million doses for domestic use.
The Seattle scientists behind the new vaccine sought to create a ‘second-generation’ COVID-19 vaccine that is safe, effective at low doses, simple to manufacture, and stable without deep freezing. These attributes could enable vaccination at a global scale by reaching people in areas where medical, transportation, and storage resources are limited.
“We know more than two billion people worldwide have not received a single dose of vaccine,” said David Veesler, associate professor of biochemistry at UW School of Medicine and co-developer of the vaccine. “If our vaccine is distributed through COVAX, it will allow it to reach people who need access.”
The University of Washington is licensing the vaccine technology royalty-free for the duration of the pandemic.
Congrats and thank you again to all R@h contributors!
6 Jul 2022, 18:27:59 UTC
· Discuss
Thank you!
We'd like to thank everyone who has contributed and continues to contribute to this project, and would like to remind everyone that there may be periods of down time while we are preparing for future large scale batches of jobs and analyzing results. We greatly appreciate your contributions which are vitally important to our ongoing research.
Thank you!
5 Nov 2020, 20:16:29 UTC
· Discuss
Coronavirus update from David Baker. Thank you all for your contributions!
Here is a short video of David Baker describing some exciting results from de novo designs targeting SARS-Cov-2.
Thank you all for your contributions to this research! Although R@h was not directly used for the work described in the publication (link provided below), R@h was used for designing relevant scaffolds. Additionally, there are currently many similar designs that bind SARS-Cov-2 and related targets that were engineered using R@h.
https://www.youtube.com/embed/ODEIN5V3yLg
More information is available from the publication, De novo design of picomolar SARS-CoV-2 mini protein inhibitors.
21 Sep 2020, 23:16:33 UTC
· Discuss
... more
News is available as an RSS feed

©2026 University of Washington
https://www.bakerlab.org