Comments/questions on Rosetta@home journal
Message boards : Rosetta@home Science : Comments/questions on Rosetta@home journal
| Author | Message |
|---|---|
|
David Baker Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 17 Sep 05 Posts: 705 Credit: 559,847 RAC: 0 |
Hope that wasn't too long a first post! looking forward to discussing issues with people here. David ID: 11592 · Rating: 0 · rate:
|
 Cureseekers~Kristof Cureseekers~KristofSend message Joined: 5 Nov 05 Posts: 80 Credit: 689,603 RAC: 0 |
I am wondering why after 3 hours, there still isn't a comment on your post, David. Everyone was screaming for this news, and now it's there, and it looks like no one want to give comments... Although I'm not a person why is known in the world of this kind of science, your post is quit clear to me. As a member of DPC, I'll surely communicate this to the rest of our team, so everyone knows it. At our team results and feedback of the project is very important. At this point I want to thank you for the effort and hope of course that you'll keep adding your comments, results, news items, etc to this thread. One remark: Maybe it's better to make that thread read-only, so we only can find your (or collegue's) posts in there. so we need more cpu power! it is kind of amazing that solving such a long standing scientific problem depends so crucially on the efforts of volunteers like yourselves! I don't know how much more cpu power it will take, but if you can each recruit ten friends or relations ... That's the spirit :-D Member of Dutch Power Cows ID: 11597 · Rating: 0 · rate:
|
|
KSMarksPsych Send message Joined: 15 Oct 05 Posts: 199 Credit: 22,337 RAC: 0 |
I just saw your post David and I'm floored. This is awesome news. What you wrote was clear, concise, and to the point (unlike most of my posts :)) I want to congratulate you and your whole team, as well as all of our crunchers here. This is going to make a huge difference to humanity. I'm just happy I get to be a part of it. Wonderful! Kathryn Kathryn :o) The BOINC FAQ Service The Unofficial BOINC Wiki The Trac System More BOINC information than you can shake a stick of RAM at. ID: 11600 · Rating: 0 · rate:
|
|
rbpeake Send message Joined: 25 Sep 05 Posts: 168 Credit: 247,828 RAC: 0 |
Hope that wasn't too long a first post! looking forward to discussing issues with people here. David I think this is a great public outreach, and may also help young people get a sense of the excitement and life of a research scientist. Hopefully it may encourage some people to pursue a career in science! :) Regards, Bob P. ID: 11602 · Rating: 0 · rate:
|
|
bruce boytler Send message Joined: 17 Sep 05 Posts: 68 Credit: 3,565,442 RAC: 0 |
Hi David, Truly amazing news. The protien prediction problem was first set forth by Linus Pauling I belive in 1935! Correct me if I am wrong. If the problem is solved through DC I belive it will go down as a major 21 century achievement. "it is clear for the still large number of proteins for which we are failing that the problem is not enough sampling, even with 100,000 independent folding runs we are not coming close enough to the native strucutre to land in its energy minimum. so we need more cpu power! it is kind of amazing that solving such a long standing scientific problem depends so crucially on the efforts of volunteers like yourselves! I don't know how much more cpu power it will take, but if you can each recruit ten friends or relations ..." If the 20 terabyte dc system is all that we end up having, given enough time, can this level of compute solve the protiens mentioned in this part of the journal post? Or is time spent on the compute not a factor and merely say another 60 terabytes is what is needed or the protiens can't be delt with? Thanks loads for the good news.....cheers 
ID: 11604 · Rating: 0 · rate:
|
|
Los Alcoholicos~Megaflix Send message Joined: 10 Nov 05 Posts: 24 Credit: 77,199 RAC: 0 |
Even for a science noob like myself it's clear information. Thanks! I'll be reading the journal regularly. ID: 11612 · Rating: 0 · rate:
|
|
BennyRop Send message Joined: 17 Dec 05 Posts: 555 Credit: 140,800 RAC: 0 |
While I read the message during its first hour online, it took awhile to make sense of a few of the comments so I could make a useful comment. Elsewhere, there were statements of 10,000 models being produced for each WU; and yet you mention needing more cpu power since 100,000 models is not enough. Am I correct in assuming that you're referring to the group of 10 or more WUs with the strange names such as "DementedOnlineSalesPitches_With_no_Fragging"? If so, would you mind describing what each of those strange names referred to - and their best score and compare that to the normal client's best score on 10,000 models. Secondly, (if we've done such) - what's the improvement we can expect from running 100,000 models instead of 10,000? (Not that there's on option at present to run everything with 100,000 models per WU.) In two places today, you've referred to David Kim as merely "David" - and in the second message, you had not previously mentioned his full name. It gets awfully confusing reading a David state, "David has been working on.." and brings to mind someone that ventures into a dark room, pulls out a ring, and starts chanting, "my presh... yusssss..." Your first post really does a great job of conveying your infectious optimism and excitement. (Although you do need to stay away from engraved golden rings.. *grin* ) Thanks. As for more users - get rid of the collection of annoying bugs so the client becomes even more reliable; reduce the bandwidth usage of the client even further, change the stats to a format that gives equal credit to all 10,000 models of a given WU, while improving the client so it can get a RMSD score of less than 3 Angstroms for a 160 molecule chain - and we shouldn't have problems attracting more cpus. (Today's mention of Stderr.txt filling 68Gigs of an 80Gig HD could explain the loss of lots of unattended machines..) ID: 11621 · Rating: 0 · rate:
|
|
Hoelder1in Send message Joined: 30 Sep 05 Posts: 169 Credit: 3,915,947 RAC: 0 |
I will definitely be a regular reader of the new journal which due to the time zone difference conveniently gets delivered right in time for breakfast (more interesting than the morning papers). Since David asked for suggestions on things to cover, here are two questions that come to mind: In his first journal report David mentioned "evolutionarily related homologs". I am guessing that homologues may be proteins where the amino acids are replaced by other, similar amino acids ? But I am completely in the dark how evolution might come into the picture. Also, assuming that such homologues are available for all proteins of interest (?), why would it be a problem to use this kind of information in Rosetta ? My second question is sort of a placeholder for my general interest in the algorithmic details and tricks employed by Rosetta: The following information appears on the 'Result' page of the new fixed-runtime WUs: # DONE :: 1 starting structures built 69 (nstruct) times # This process generated 69 decoys from 69 attempts The number in front of (nstruct) seems to be the number of structures that are determined within the specified run time, but what are decoys and attempts ? I have seen rare cases where the number of attempts was _much_ larger than the number of structures. Anyway, I probably could think of many more questions should David run out of things to discuss, but I have the impression that he will know best what will be suitable for the journal. I am very much looking forward to the future installments of the new Rosetta (breakfast-) journal/seminar/lecture... ID: 11678 · Rating: 0 · rate:
|
|
Robert Everly Send message Joined: 8 Oct 05 Posts: 27 Credit: 665,094 RAC: 0 |
Thanks for the news. While 99.9% of the science info is over my head, it is nice to know that progress is being made. ID: 11685 · Rating: 0 · rate:
|
|
rbpeake Send message Joined: 25 Sep 05 Posts: 168 Credit: 247,828 RAC: 0 |
This may be too personal, or not of interest to others or to David (and I completely respect that), but as a thought I toss it out: David, How did you become interested in pursuing a career in science, and how did you end up doing what you are doing? Not too personal, but I find the journey to what one is doing today interesting, and perhap might be inspiring to younger people seeking a future path. Regards, Bob P. ID: 11686 · Rating: 0 · rate:
|
|
hugothehermit Send message Joined: 26 Sep 05 Posts: 238 Credit: 314,893 RAC: 0 |
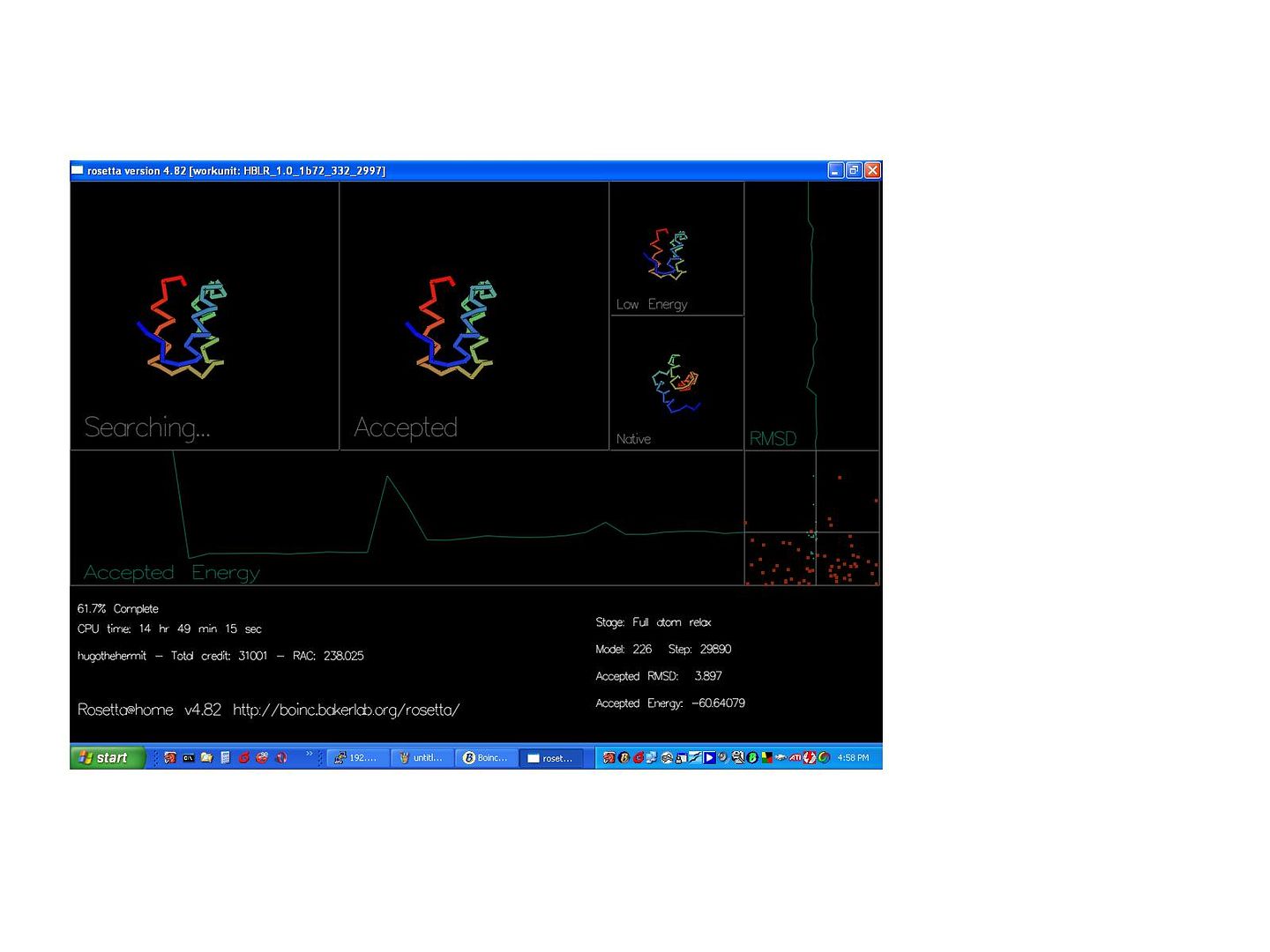
Must say Dr D.B great journal, I log on just to read it, as I'm doing now, no results to report or anything :). On reading your journal I've decided this truly seems to be a solvable problem, when someone said something about 500 dimentions and thought about all those atoms and electrons I must admit I thought "this isn't happening in my life time", but I now I'm not that sure, it seems that brains (you and your team) working with brawn (us) may actually make this giant leap. Truly amazing. Now a question for you: In this work unit, I have the minimum RMSD by the look of it, but too high an energy. I know that you had both the minimum energy and the minimum RSMD in the stats, I was wondering why does the RMSD matter at all, should a protein be both the smallest it can be and have the least energy? Or should it just have the least energy? Is RMSD just a short cut to working out the energy? BTW congrats to, I assume, Jack. I'm not sure if anybody else has noticed but the ribbons into pipes in the graphics was a nice touch that didn't go unnoticed ;). ID: 11703 · Rating: 0 · rate:
|
|
uioped1 Send message Joined: 9 Feb 06 Posts: 15 Credit: 1,058,481 RAC: 0 |
The homolog insight is truly a leap forward. That is the sort of insight that comes along rarely and can be quite beneficial to search problems of this magnitude! Congratulations! I wonder if it is required that the homologs be evolutionarily related, or if we could generate homologs that would serve the same purpose for the search. This might have the advantage of strengthening the value of the results (just like using the RMSD as a heuristic would invalidate them) In answer to hugo the hermit: [quote}In this work unit, I have the minimum RMSD by the look of it, but too high an energy. I know that you had both the minimum energy and the minimum RSMD in the stats, I was wondering why does the RMSD matter at all, should a protein be both the smallest it can be and have the least energy? Or should it just have the least energy? Is RMSD just a short cut to working out the energy?[/quote] RMSD is a measure of the difference between the two foldings of a specific protien. I'd venture an educated guess that it stands for "[square]Root Mean Squared Deviation" For the runs we've done thus far, we've known the natural folding so we can calculate the RMSD and use it to evaluate how well the algorithm works. For the application to be usefull, however, it can't look at the RMSD to decide how to fold the protien, because we can't calculate it for protiens whose natural structures are unknown. I'm not a chemist, so I can't state with 100% certainty, but I think that the natural structures will have the lowest energies possible, thus in some sense we are trying to get to where we can use the energy of a structure as an approximation of the rmsd. ID: 11738 · Rating: 0 · rate:
|
|
hugothehermit Send message Joined: 26 Sep 05 Posts: 238 Credit: 314,893 RAC: 0 |
From uioped1 RMSD is a measure of the difference between the two foldings of a specific protien. I'd venture an educated guess that it stands for "[square]Root Mean Squared Deviation" For the runs we've done thus far, we've known the natural folding so we can calculate the RMSD and use it to evaluate how well the algorithm works. For the application to be usefull, however, it can't look at the RMSD to decide how to fold the protien, because we can't calculate it for protiens whose natural structures are unknown. I'm not a chemist, so I can't state with 100% certainty, but I think that the natural structures will have the lowest energies possible, thus in some sense we are trying to get to where we can use the energy of a structure as an approximation of the rmsd. Thanks for the infomation, I seem to remember reading somthing like that some time ago (I'd forgotten), so I'm sure you are right. But that begs the questions (now I'm not sure how low the RMSD is on the WU picture I posted as I wasn't looking at it at the time but it was pretty low). Why, when the ab initio is that close to the original is the energy so high (does the full atom relax need tweaking)? Or is just a little out (ab initio) energetically large? ID: 11747 · Rating: 0 · rate:
|
|
David Baker Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 17 Sep 05 Posts: 705 Credit: 559,847 RAC: 0 |
From uioped1 the energy can be high even a little bit a way from the correct solution because small displacements can put atoms too close to each other which is very costly energetically. ID: 11748 · Rating: 0 · rate:
|
|
BennyRop Send message Joined: 17 Dec 05 Posts: 555 Credit: 140,800 RAC: 0 |
the energy can be high even a little bit a way from the correct solution because small displacements can put atoms too close to each other which is very costly energetically. After the client hits a fairly low RMSD - have you tried adjusting each atom in the chain one at a time - for even lower energy placements? Calculate its energy some tiny distance up, down, in, out, right, left. Move it to the location with the lowest energy, and then repeat another 9-19 times? And then move on to the next atom in the chain. Kind of like a zipper.. lock one set into place and move onto the next set of teeth. Or is it better to start in the middle of the chain and work out to both ends of the chain? ID: 11750 · Rating: 0 · rate:
|
|
David Baker Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 17 Sep 05 Posts: 705 Credit: 559,847 RAC: 0 |
the energy can be high even a little bit a way from the correct solution because small displacements can put atoms too close to each other which is very costly energetically. we do something like this-we move everything in the direction of the steepest decrease in the energy (by computing the derivative of the energy with respect ot all of the degrees of freedom and then moving in this direction) ID: 11769 · Rating: 0 · rate:
|
|
thom217 Send message Joined: 29 Oct 05 Posts: 12 Credit: 182 RAC: 0 |
Hi Dr. Baker, I think that if you keep informing people there is at least one or two proteins you are folding that have a direct link to some disease it will help to bring more people to Rosetta. Another idea to try is to create a GPU client for Rosetta. Thanks. ID: 11778 · Rating: 0 · rate:
|
|
Dimitris Hatzopoulos Send message Joined: 5 Jan 06 Posts: 336 Credit: 80,939 RAC: 0 |
I think that if you keep informing people there is at least one or two proteins you are folding that have a direct link to some disease it will help to bring more people to Rosetta. Perhaps someone else can explain it better (or correct me if I'm wrong), but until then, let me try: Rosetta's main goal is to eventually identify UNKNOWN proteins, i.e. those which 3D shape and thus biological function is currently unknown. Only after the determination of 3D shape has been performed, can biologists and biomed scientists study those shapes to decide that e.g. protein X is related to disease Y. E.g. HPF2 project run at IBM's World Community Grid is applying Rosetta software (the one that we help develop here) to "cancer biomarkers" (proteins found at dramatically increased levels in cancer tissues), human secreted proteins and malaria. (see HPF update 16-jan-06 Here we're helping improve the algorithms used by projects such as HPF. Here's some more (from HPF): "The Human Proteome Folding project is basic medical research. We are given some fundamental components of cells (proteins of unknown function) and we try to deduce their shape, then from this deduce which other proteins they interact with, and how. It is like pouring the components of an "Erector set" (editor: construction toy) onto the living room floor and trying to figure out what goes with what. The goal is to figure out the functional networks that drive basic cell processes. Once you have identified the function of a protein you can: 1. Select it as a target for a drug to interfere with its function 2. Figure out how it works and design a drug to duplicate the effect 3. Develop a diagnostic test to detect the concentration of that protein in order to measure the level of activity Without the protein information, these three things are very important objectives that can only be accomplished by mass screening of a vast number of chemical compounds, hoping for a lucky breakthrough. Even with this information, a great deal of work, skill and luck is required to develop a drug. The HPF project can provide very useful information for drug development, but it is aimed at basic understanding that can then be used to develop drugs. We are providing the shape information. Scientists studying the databases with this structural information will predict the function of the proteins (annotate the proteins)." Have a look at the URL in my sig, I provide many links with info about the various life-science projects. Best UFO Resources Wikipedia R@h How-To: Join Distributed Computing projects that benefit humanity ID: 11799 · Rating: 0 · rate:
|
|
hugothehermit Send message Joined: 26 Sep 05 Posts: 238 Credit: 314,893 RAC: 0 |
(2). I'm a bit disappointed that the total cpu power has remained constant for the past weeks rather than increasing as it had up until recently. More users and hosts are joining every day, but this is not translating into increased computing capability. We aim to extend our calculations to larger proteins based on teh success we ahve been having with proteins under 100 amino acids, but this will require a significant increase in computing power. Please let me know if there is anything I can do to help with recruiting Dr D.B I've sent a request to the Australian Broadcasting Corporation (ABC, it's the Australian version of BBC or PBS (?) ), for a documentary on Rosetta@Home. To try to increase the participants in the project. Like all publiclly funded TV/Radio stations it's short of money so I can't promise anything, but even if they just do a radio programme it could make a differance. The way I see it is that 1000 users = about 1 million dollars worth of hardware, so every bit helps. I just thought I should let you know, so that if anybody bothers ringing or e-mailing you form ABC Australia you have a chat with them :). Ps I'm going to send you then request, so don't worry about getting an e-mail from hugothehermit.XXXXX it's just me ;) Edit: I should at least be able to spell Australia you'd think :) ID: 11813 · Rating: 0 · rate:
|
|
carp Send message Joined: 4 Jan 06 Posts: 12 Credit: 599,555 RAC: 0 |
Recruitment Ideas. Send a letter and information packet to CEOs', CFOs', I.T officers of all the health insurance companies(Life, etc), and these companies cracking down on lifestyle choices of their employees. Any of them that can't find a way to get you some CPU power are just hypocrits. They are always tooting the horn of lowering health insurance costs through better habits, excercise, diet, excercise, etc. So giving up some CPU to find new, better, and cheaper therapies should make a nice fit for them and give them another reason to toot their own horn. I work in a company of over 2500. Many of the machines are idle most the day and left on at night. Even 10% of total CPUs is something. And yes it is one of those target companies. I just haven't had any luck with getting them on the bandwagon. ID: 11823 · Rating: 0 · rate:
|
{kind=link}
Message boards :
Rosetta@home Science :
Comments/questions on Rosetta@home journal

©2026 University of Washington
https://www.bakerlab.org