Posts by Mark
|
21)
Message boards :
Cafe Rosetta :
proteins----- protein research
(Message 78137)
Posted 18 Apr 2015 by Mark Post: http://www.powerplantjobs.com/ppj.nsf/powerplants1?openform&cat=ny&Count=500 Not sure this is the right link?! |
|
22)
Message boards :
Number crunching :
Metrics
(Message 77970)
Posted 23 Feb 2015 by Mark Post: (No idea why the picture is not displaying, tags are correct...?) Thx Timo, good catch! |
|
23)
Message boards :
Number crunching :
Metrics
(Message 77966)
Posted 23 Feb 2015 by Mark Post: The lack of a trend line for increased decoy count for a larger protein could also be an accounting difference. As was discussed previously in the thread, some proteins are studied in phases and so from start to finish perhaps data for such a protein appears as 4 or 5 separate runs. I have the sequence data for the proteins and they are all unique.... |
|
24)
Message boards :
Number crunching :
Metrics
(Message 77962)
Posted 22 Feb 2015 by Mark Post: Yep thx for that. I knew it was realistically incalculable so I didn't bother to find out by how much! I plotted a graph from the data, and there doesn't seem to be much correlation between protein size and total run duration. This may well be a factor in how the runs are initialised, if a person is estimating how many decoys will be needed for example. Anyway this is what it looks like 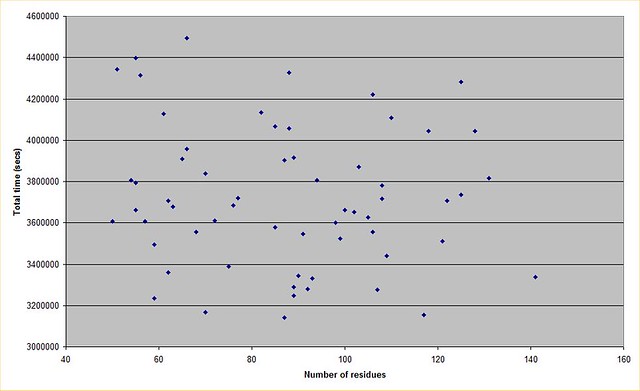 https://farm9.staticflickr.com/8637/16610904862_fb4cd398e2_z.jpg (No idea why the picture is not displaying, tags are correct...?) |
|
25)
Message boards :
Number crunching :
Metrics
(Message 77936)
Posted 13 Feb 2015 by Mark Post: Are you looking for decoy sets? If you are looking for runtime benchmarks/data, I believe the decoy sets have runtime info for each decoy, which of course are dependent on the machines that produced them. Hi David, Thx for your reply and your time. Not sure exactly what you mean by decoy sets. Do you mean all the decoys generated for one sequence? Sure that would be interesting, but not essential. I am looking for say 3 protein input/starting sequences of approx 40, 90 and 150 AA length. I presume you start the fold with the residues in a straight long chain? I then just need the total cpu time that R@H contributors spent on it, and number of decoys produced before you got to at least one decoy within 2Å RMSD. (Obviously I could extract the runtime from each decoy and sum if necessary) I'm also unsure of the end criteria R@H uses, where you decide when a sequence is "solved" and you move onto the next? I would guess you estimate the number of decoys required, run that, and then find out if it actually gave a good decoy or not and then maybe repeat if it is an interesting target, or do you do something different? If you could upload those sequences and data somewhere I would be very grateful! Definitely a beer or two for you if you come to London! :) Thx Mark |
|
26)
Message boards :
Number crunching :
Metrics
(Message 77931)
Posted 13 Feb 2015 by Mark Post: Rosetta is developed by numerous academic institutions through the Rosetta Commons. There are continuous unit tests, performance benchmarks, etc.. run after each code checkin. The unit and performance tests framework is part of the code distribution. The code is constantly being developed. In fact there's a developers meeting going on right now in Boulder, CO where many developers from the various commons institutions meet to talk about code development, testing, optimization, etc. There's also an annual meeting to talk about the science. Hi David, yes I'm am focusing on ab initio folding. Are the benchmarks and more specifically, the results available somewhere? I had a quick look through the code distribution and I see the framework to run standardised tests, I see some small test protein cases, but if we are talking 56 days for repeated tests to get to 2Å RMSD, I think I prefer to use your data! :) |
|
27)
Message boards :
Number crunching :
Metrics
(Message 77926)
Posted 12 Feb 2015 by Mark Post: No, I'm not aware of any aggregated data on runtimes. See, this is what strikes me as strange. How do you know if new techniques are working OK without measurement? How do you know where bottlenecks and development areas are without measurement? How do I know if an idea I have is better or worse than the current state of play without some kind of metric to measure against? By making performance data available, you would be encouraging new ideas and development of Rosetta as a whole, to the benefit of everyone. The type of thing initially required is so straightforward that it must surely be available somewhere by someone in the R@H program...? What needs to be done to get hold of it? Who do I need to contact? Thx |
|
28)
Message boards :
Number crunching :
Metrics
(Message 77921)
Posted 12 Feb 2015 by Mark Post: Definitely not sensitive info :) I would guess the 56 cpu days is spread over many computers hence they would get results in under 24 hours.... |
|
29)
Message boards :
Number crunching :
Metrics
(Message 77917)
Posted 11 Feb 2015 by Mark Post: Definitely not sensitive info :) Ah brilliant, that's the kind of thing I was looking for! So 56 cpu days is a rough ballpark for a 100 residue protein (with wide variation!) :) Thx also to TJ. Yes I appreciate there is a large range of "difficulty of solving", maybe I should have used the term "median" rather than "average"! Does anyone happen to have say 3 test proteins with solving stats that I could play with? I am currently using a protein that was sent out to Rosetta@Home some time in Oct (fasta:ACSGRGSRCPPQCCMGLRCGRGNPQKCIGAHEDV) which I think I pulled out of the software correctly! To get some solving stats for it would be great. Looking at the stats you mention, the cpu time is the time for that job on my PC, although the same job (with different seeds presumably) will have been sent to other PCs as well? Is there any way to see them (the job name is not hyperlinked) and hence give the the total time (over all PCs) for that protein? Also I note the stderr doesn't give the residue count so unless I grab each protein myself, I won't have that data either.... Thx to all for pointing me in the right direction! Mark |
|
30)
Message boards :
Number crunching :
Metrics
(Message 77912)
Posted 11 Feb 2015 by Mark Post: The "Sampling bottlenecks in de novo protein structure prediction" paper provides some insight. OK thx for that. I see it gives a rough figure of say 1000-10000 samples to get an acceptable RMSD (for an approx 100 residue protein)[Fig 3], and how using specific constraints cuts that down. However the paper is about 6 years old and obviously a) Rosetta has moved on since then and b) computing speed has moved on since then through Moores law. Also the paper didn't mention (that I noticed) actual time taken for each of those samples to finish, obviously a factor. I take the point the analysis is done in stages too. However I still don't know how many compute hours to get to a 2Å RMSD decoy for an "average" 100 residue protein...? Is much performance testing done? I would think someone has the Rosetta@Home figures for how many decoys are sent out for a given protein and how long they take? Is the information sensitive in some way? Thx |
|
31)
Message boards :
Number crunching :
Metrics
(Message 77906)
Posted 9 Feb 2015 by Mark Post: Some proteins are studied in phases, where results from one phase are used to place some constraints on the search space of subsequent phases. So I'm sure there is no simple answer to your question. Thx for that. I have tried foldit and do have a sense of the difficulty. Does anyone have any actual hard data though on the amount of compute required for a particular protein? |
|
32)
Message boards :
Number crunching :
Metrics
(Message 77903)
Posted 8 Feb 2015 by Mark Post: Hi, I was interested in the metrics/efficiency of the search as a research area. My understanding is that Rosetta@Home randomly picks a start point then uses simulated annealing to find a minima. Multiple decoys are produced from multiple computers and plotted on a graph such as this. (In this example it shows a nice funnel with the folds of interest towards the turquoise area in bottom left)  My question is how many data points are generated per protein investigation and how much CPU time is required to produce one point? Obviously larger proteins with more residues are going to take longer/more effort. Is there a rough relationship between number of AA to search effort? Thx Mark |
|
33)
Message boards :
Rosetta@home Science :
protein-protein docking at Rosetta@Home
(Message 77718)
Posted 4 Dec 2014 by Mark Post: Still no real updates here? No news seems to indicate an aloofness to us mere mortals. I know what you mean, however it could also be that they're really busy doing science.... |
|
34)
Message boards :
Number crunching :
GPU Potential
(Message 77633)
Posted 9 Nov 2014 by Mark Post: From my standpoint, this project has potential with a GPU app. BOINC regularly polls this project trying to find tasks for my Nvidia, even though I don't see any GPU apps. From what I know, the way data is analyzed and processed, I think a GPU would be able to process tasks much faster than a CPU. There's plenty of RAM to hold the process, and the multiple cores would allow for more data to be sent through. How hard would it be to implement a GPU version of Rosetta@home? This is a question that gets regularly asked. The answer is very hard and not top of the priorities |
|
35)
Message boards :
Rosetta@home Science :
Folding of known structures
(Message 77626)
Posted 2 Nov 2014 by Mark Post: Hi Mark et al. Ah thx Chris, that makes sense! Yes in that case it is the "known" solution that is confusing. Thx again for clarifying Mark |
|
36)
Message boards :
Rosetta@home Science :
Folding of known structures
(Message 77595)
Posted 19 Oct 2014 by Mark Post: I am just a volunteer and not in the BakerLab, but in very general terms, if you were to develop an algorithm that is to predict structures, and you test it by creating a pile of possible models of things of unknown structure... then what have to learned about how to improve your algorithm? Running it against known structures is how you prove your algorithm is working well, or came up with the same correct answer, or an answer with even better precision, or using dramatically less compute power. Yes I get that point, but as I said in the original post it seems about 70% of the time you are folding to a known solution. If you multiply that up over the number of participants that's an awful lot of testing data. You would then be able to make lots of changes to the algorithms as you have lots of data to go on. However the minirosetta program gets updated infrequently, which brings me back to the original question.... |
|
37)
Message boards :
Rosetta@home Science :
Folding of known structures
(Message 77592)
Posted 19 Oct 2014 by Mark Post: I am curious about this as well. When in the screensaver it has 4 boxes and the fourth one is called "native", the native one is the actual solution probably derived from xray crystallography. Also you have a RMSD figure/graph which is the "difference" between the native structure and the structure the computer has just folded |
|
38)
Message boards :
Rosetta@home Science :
Folding of known structures
(Message 77582)
Posted 17 Oct 2014 by Mark Post: Hi, I was wondering why there is so much folding being done of structures with a known native solution? I could understand that it is useful sometimes as a metric to test the folding capabilities of Rosetta, and to develop new efficient folding movers to include in Rosetta, but it seems that 60-70% of the WU I get are folding to a known solution which seems awfully high. The only other thing I can think of is that you are trying to find the sequence for a computed binder structure. Is that it? |
|
39)
Message boards :
Rosetta@home Science :
Some new challenges for the team?
(Message 77565)
Posted 8 Oct 2014 by Mark Post: Hi, I know David is always asking for new challenges and areas of research, so how about having a crack at the Longitude Prize? http://www.longitudeprize.org/challenge/antibiotics The challenge is " to create a cheap, accurate, rapid and easy-to-use point of care test kit for bacterial infections." "Point-of-care test kits will allow more targeted use of antibiotics, and an overall reduction in misdiagnosis and prescription. Effective and accurate point of care tests will form a vital part of the toolkit for stewardship of antibiotics in the future. This will ensure that the antibiotics we have now will be effective for longer and we can continue to control infections during routine and major procedures. " So a simple set of specific protein binders on a chip/wells/card of some sort seems to meet the requirements. Oh, and there's a $15 million prize....! Failing that, how about MRSA, C Diff, TB or African Sleeping Sickness binders? |
|
40)
Message boards :
Rosetta@home Science :
Rosetta@home Research Updates
(Message 77563)
Posted 8 Oct 2014 by Mark Post: Any new updates on research? Thx |
Previous 20

©2024 University of Washington
https://www.bakerlab.org